ARES简介
Ab initio atomic mateRial modEling Software at JLU(ARES)是一款基于密度泛函理论,利用实空间/平面波基组结合赝势方法进行电子结构计算的第一性原理软件,可以进行原子尺度的电子结构计算和分子动力学模拟,可跨节点多核运算,支持超大规模集群并行。ARES可根据模拟体系的大小,自动选取计算效率最优的展开基组,在64核单节点上即可实现3000电子以上体系的快速模拟。
主要功能特点
- 基本的电子结构计算模拟
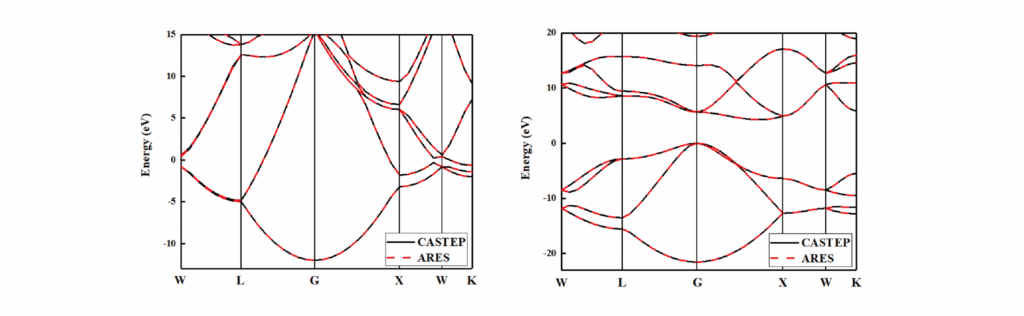
能带计算
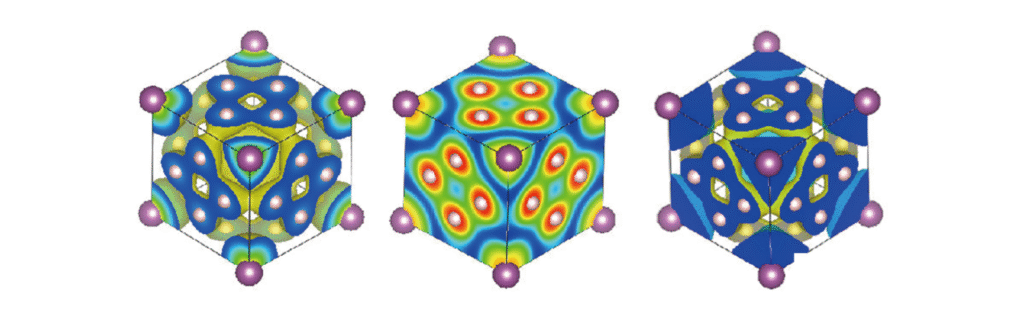
电子自洽
结构弛豫
- 复杂边界条件的材料性质计算
晶体(周期性)
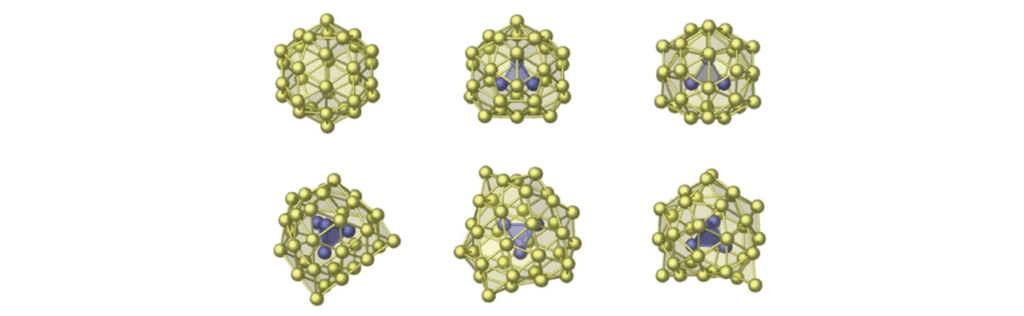
团簇(非周期性)
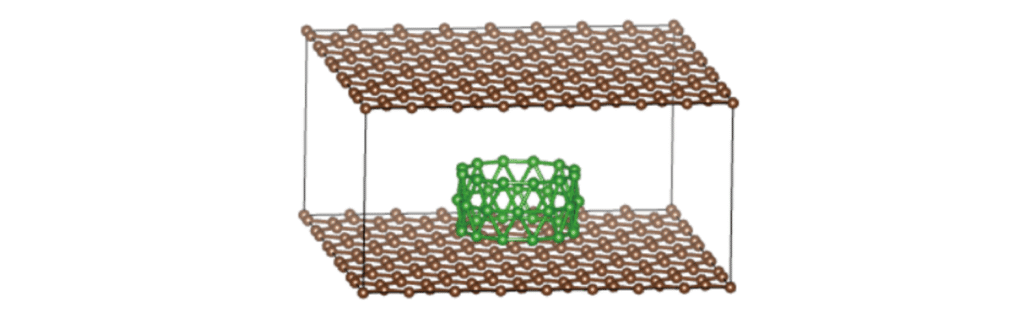
复杂体系(半周期性)
- 机器学习加速

分子动力学模拟
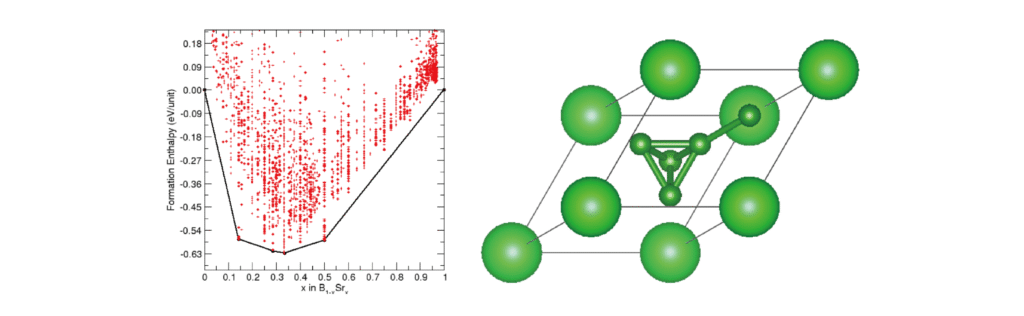
结构预测
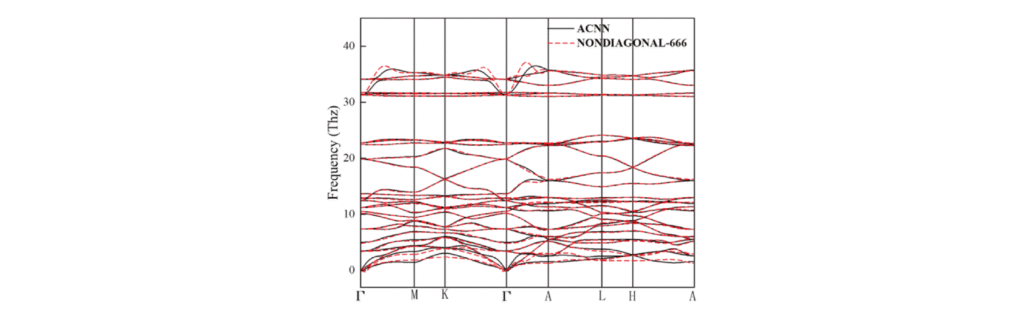
声子谱
典型应用
基于ARES的原子截断方法,对BSb体系的纳米管进行电子结构计算,给出了随纳米管孔径增大带隙的变化情况,原胞结构原子数4-88,计算结果表明BSb纳米管是潜在的半导体材料,且形成能的计算结果表明,该纳米管的合成较碳纳米管更为容易。
固态电解质是全固态电池的核心部件,Na5YSi4O12是一类具有高离子电导率的钠离子固态电解质,对其性质的理解缺少原子尺度的研究。我们首次使用机器学习势辅助的分子动力学对Na5YSi4O12的电化学性质进行了模拟,阐明了结构框架中离子输运的基本模式。对小尺寸(132原子)和大尺寸(132000原子的模拟)计算出的离子电导率与实验观测到的数值一致,证明机器学习势方法的可靠性。
我们提出了一种以合成的sp3碳结构为原型设计sp3杂化硼化物超导体的策略。我们进一步对低于100 GPa的Sr-B系统进行了系统结构预测,以验证Sr2B5是否热力学稳定。相图表明,超导Sr2B5在38-44GPa下热力学稳定,在40 GPa下Tc高于100 K,有可能保持在均匀的环境压力下,在所有已知的热力学稳定的金属硼化物中创造了新的超导记录。